FOLLOW-UP FREQUENCY IN ILD

FOLLOW-UP FREQUENCY IN ILD - Nurse connect
Several interstitial lung diseases (ILDs) may become progressive at any time in the disease course. Once a patient is diagnosed with ILD, close monitoring is important to identify progression early. It is estimated that it generally takes 9-12 months to detect progressive fibrosis after it develops.1 Delays in the diagnosis of progressive fibrosis can significantly decrease the life expectancy of patients.2 Close and frequent monitoring of ILD allows early identification of progression and enables early treatment. This is important to slow down the loss of lung function as much as possible.2-6
HOW OFTEN TO MONITOR?
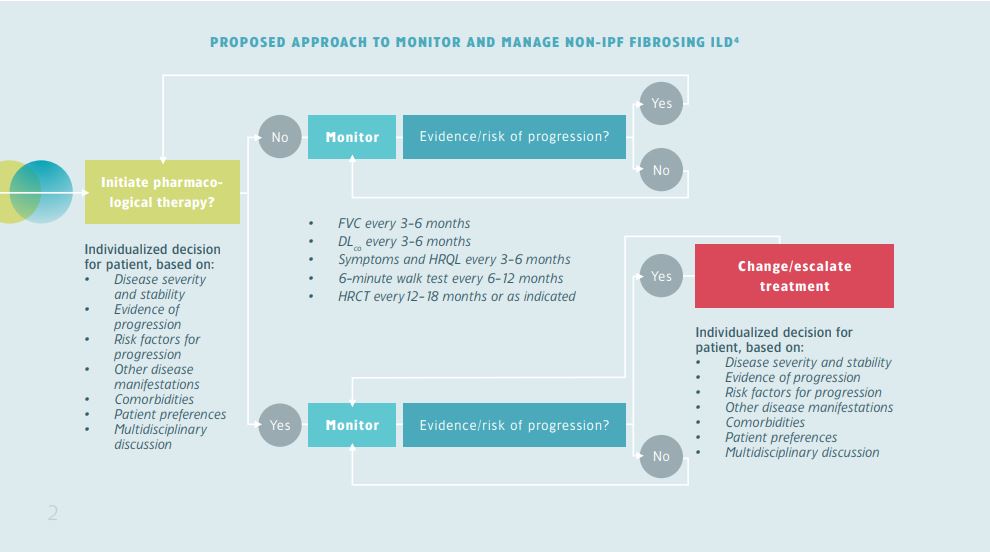
For most ILDs there are no guidelines on how frequently patients should be monitored, although recommendations exist. The type and frequency of monitoring is decided in a multidisciplinary discussion as it is highly dependant on individual factors, such as disease severity, evidence of progression, risk factors for progression, and comorbidities.7 These factors could help to inform monitoring strategy and frequency.
Non-IPF fibrosing ILD
For patients with a fibrosing ILD that is not idiopathic pulmonary fibrosis (IPF), it is proposed to perform lung function tests (FVC and DLco), evaluate symptoms and assess health-related quality of life (HRQL) every 3-6 months.7 In addition, many providers will perform high-resolution computed tomography (HRCT) at the patient’s initial presentation and then every 12-18 months to detect if the fibrosis is progressive. The 6-minute walk test may be performed every 6-12 months to support evidence for decline in exercise capacity.
IPF
IPF is by definition a progressive fibrosing ILD, and prompt initiation of antifibrotic therapy is necessary. To monitor IPF, the official ATS/ ERS/JRS/ALAT Clinical Practice Guideline recommends to consider:
- pulmonary function tests and the 6-minute walk test every 4-6 months, or sooner if clinically indicated
- HRCT if there is concern for an acute exacerbation
- annual HRCT if there is suspicion of clincial worsenening or risk of lung cancer.8
|
SSc-ILD
Experts in the management of patients with systemic sclerosis-associated ILD (SSc-ILD) have proposed that patients should be monitored for disease progression every 3-6 months using multiple methods, including symptom assessment, pulmonary function tests, exercise-induced blood oxygen desaturation and, where appropriate HRCT.9,10
RISK FACTORS
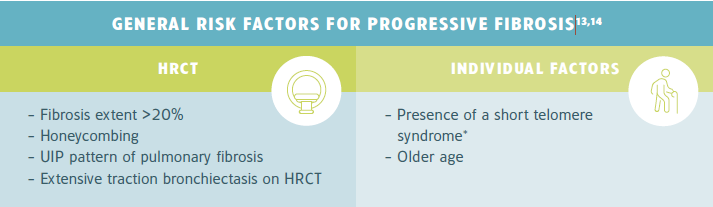
Risk factors for ILD progression could help to inform monitoring frequency. For example, an older male patient with rheumatoid arthritis-associated ILD (RA-ILD) who has a usual interstitial pneumonia (UIP) pattern on HRCT and a FVC of 60% may be monitored every 3 months rather than every 6 months.11,12
|
*See the back page for further information on short telomere syndrome.
CTD-ILD
In connective tissue disease-related ILD (CTD-ILD), several autoantibodies have been associated with a greater risk of ILD progression, such as anti-topoisomerase I (anti-Scl-70) in patients with early systemic sclerosis.11,12

|
REFERENCES
REFERENCES
-
Wijsenbeek M, Kreuter M, Olson A, et al. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35(11):2015-24.
-
Cosgrove GP, Bianchi P, Danese S, et al. Barriers to timely diagnosis of interstitial lung disease in the real world: the INTENSITY survey. BMC Pulm Med. 2018;18(1):9.
-
Cottin V, Hirani NA, Hotchkin DL, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
-
Walsh SL, Devaraj A, Enghelmayer JI, et al. Role of imaging in progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180073.
-
Roofeh D, Jaafar S, Vummidi D, et al. Management of systemic sclerosis-associated interstitial lung disease. Curr Opin Rheumatol. 2019;31(3):241-9.
-
Wells AU, Flaherty KR, Brown KK, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases—subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 2020;8(5):453-60.
-
Nambiar AM, Walker CM, Sparks JA. Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians. Ther Adv Respir Dis. 2021;15:17534666211039771.
-
Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47.
-
Hoffmann-Vold A-M, Maher TM, Philpot EE, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. The Lancet Rheumatology. 2020;2(2):e71-e83.
-
Perelas A, Silver RM, Arrossi AV, et al. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. 2020;8(3):304-20.
-
Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med. 2020;383(10):958-68.
-
Bosello SL, Beretta L, Del Papa N, et al. Interstitial Lung Disease Associated With Autoimmune Rheumatic Diseases: Checklists for Clinical Practice. Front Med (Lausanne). 2021;8:732761.
-
De Sadeleer LJ, Goos T, Yserbyt J, et al. Towards the Essence of Progressiveness: Bringing Progressive Fibrosing Interstitial Lung Disease (PF-ILD) to the Next Stage. J Clin Med. 2020;9(6):1722.
-
George PM, Spagnolo P, Kreuter M, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. 2020;8(9):925-34.
-
Molina-Molina M, Planas-Cerezales L, Perona R. Telomere shortening in idiopathic pulmonary fibrosis. Acortamiento de los telómeros en fibrosis pulmonar idiopática. Arch Bronconeumol (Engl Ed). 2018;54(1):3-4.
* Telomeres are repeated sequences of DNA at the ends of chromosomes that protect them from degradation. Telomeres gradually shorten in each cell division, which eventually cause the cell to permanently shut down (senescence) or go into apoptosis. Some mutations may cause accelerated telomere shortening, which is associated with premature aging and pathologies that involve abnormal tissue repair.15
Telomere shortening is described in 25% of IPF patients and in over 50% of patients with familial IPF. IPF patients with telomere shortening, particularly those with familial IPF, generally have a worse prognosis and more morbidity after lung transplantation.15
Related Items

ILD-sjuksköterskors kompetenser och profil

Övervakning av ILD-progression: vilket test ska utföras och när?
