MONITORING FOR ILD PROGRESSION:
WHICH TEST TO CONDUCT AND WHEN?

Monitoring for ILD Progression: which tests to conduct & when - Nurse connect
TIME IS OF THE ESSENCE
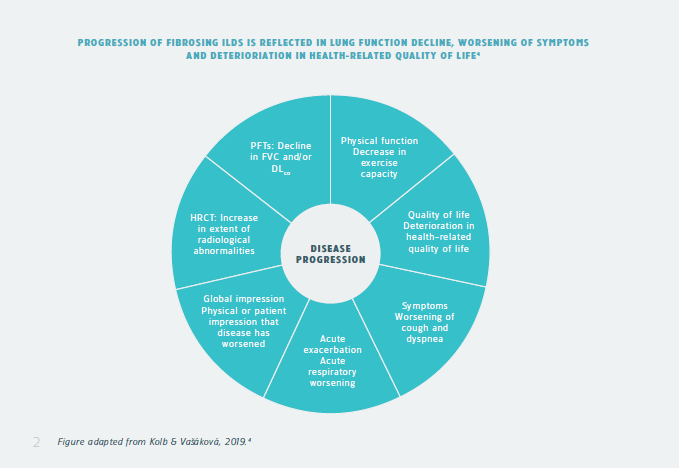
Many patients with interstitial lung disease (ILD) can develop a progressive phenotype at any time in their disease course. Progressive ILD is associated with worsening lung function, respiratory symptoms, quality of life, and ultimately death from respiratory failure.1
Fibrosis in ILD is often irreversible, so for patients it is of vital importance to receive treatment as early as possible and slow down lung function decline.1,2
As such, patients who are diagnosed with ILD should be regularly monitored for ILD progression.
How is ILD progression defined?
In a recent guideline, a multidisciplinary committee addressed progression of pulmonary fibrosis in patients with ILDs other than idiopathic pulmonary fibrosis (IPF).3
Progressive pulmonary fibrosis (PPF) is defined as at least two of the three criteria in the table on the next page, occuring within the past year with no alternative explanation.3
|
PPF criteria

|
WHICH TESTS TO CONDUCT?
Once a patient is diagnosed with ILD, close monitoring becomes important to detect changes in the disease course. Monitoring for progressive ILD should involve regular assessment of physiology (including pulmonary function tests), symptoms, and, when appropriate, high-resolution computed tomography (HRCT).5
TESTS TO CONDUCT

Other considerations when monitoring for ILD progression

|
*The 6MWD may be more challenging in patients with rheumatic diseases due to the impact of extra-pulmonary complications5
FOLLOW-UP
Assess needs by conversation
Besides tests to detect disease progression, patients’ needs for additional care may change throughout the disease course. ILD impacts the lives of patients in many ways and patients may have needs for additional support, including psychological, physical, spiritual, social and cultural needs.7
Nurses spend more time with patients and their families than any other healthcare professional. They know the values and needs of patients, the problems they face in their daily lives and are able to recognize changes in patients’ health status.8
|
For more information on what palliative care entails and how to start conversations to identify palliative care needs, follow the e-learning Palliative care in interstitial lung diseases – Scope and Conversations
IPF
Idiopathic Pulmonary Fibrosis (IPF) is by definition a progressive fibrosing ILD. Patients with IPF should immediately start with antifibrotic therapy upon diagnosis.9
In follow-up appointments for people with IPF10:
- Assess lung function
- Assess for oxygen therapy
- Assess for pulmonary rehabilitation
- Offer smoking cessation advice, in line with Smoking cessation services (NICE public health guidance10)
- Identify exacerbations and previous respiratory hospital admissions
- Consider referral for assessment for lung transplantation in people who do not have absolute contraindications
- Consider psychosocial needs and referral to relevant services as appropriate
- Consider referral to palliative care services
- Assess for comorbidities (which may include anxiety, bronchiectasis, depression, diabetes, dyspepsia, ischaemic heart disease, lung cancer and pulmonary hypertension).
- Give information on patient support groups
WHEN TO MONITOR?
Non-IPF fibrosing ILDs
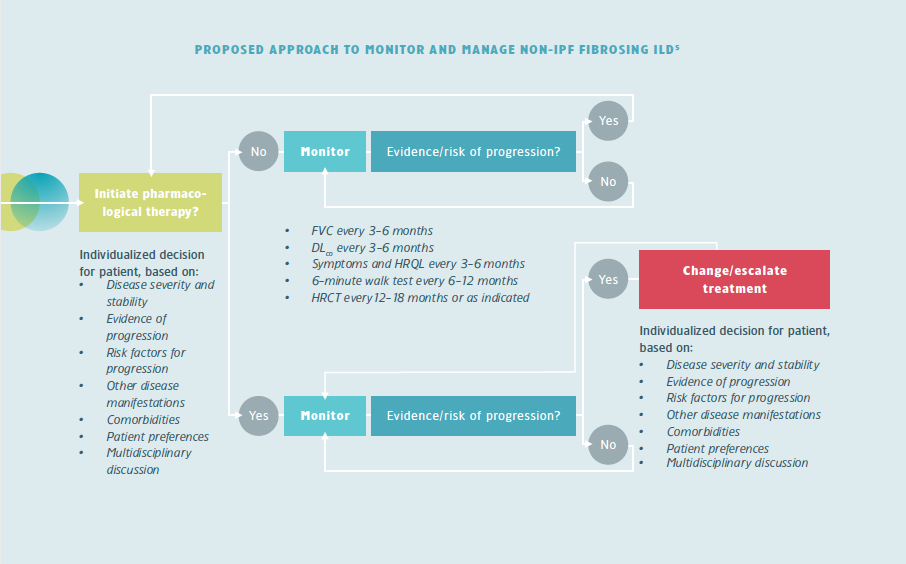
For most ILDs there are no guidelines on how patients should be monitored. Monitoring should be based on an individualized approach that involves regular assessment of different factors.5
At initiation of anti-fibrotics in progressive fibrosing ILDs, information could be provided by the ILD nurse. A (telephone) consultation may be scheduled after 2-6 weeks to e.g. check for side effects and treatment adherence. After that the patient is monitored regularly for disease progression.
IPF
The National Institute for Health and Care Excellence (NICE) recommend to follow-up patients with IPF every 3 months or sooner if they are showing rapid disease progression, every 6 months or sooner if they have steadily progressing disease or initially every 6 months if they have stable disease and then annually if they have stable disease after 1 year.10
The official ATS/ERS/JRS/ALAT Clinical Practice Guideline recommends to consider:
- pulmonary function tests and the 6-minute walk test every 4-6 months, or sooner if clinically indicated
- HRCT if there is concern for an acute exacerbation
- annual HRCT if there is suspicion of clincial worsenening or risk of lung cancer.3
SSc-ILD
Experts in the management of patients with systemic sclerosis-associated ILD (SSc-ILD) have proposed that patients should be monitored for disease progression every 3-6 months using multiple methods, including symptom assessment, pulmonary function tests, exercise-induced blood oxygen desaturation and, where appropriate HRCT.11,12
Home monitoring
Digitisation and the COVID-19 pandemic has accelatered the use of home spirometry and encouraged studies with other devices, such as pulse oximeters, activity trackers and cough monitors. Home monitoring may facilitate better collaboration between patients and healthcare providers, potentially leading to improved quality of life for patients. However, the widespread use of home monitoring may be challenging due to technical, analytical and implementation issues.13
EVIDENCE-BASED CONSENSUS STATEMENT FOR MANAGEMENT OF SSC-ILD11

|
REFERENCES
-
Cottin V, Hirani N, Hotchkin D, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
-
Wijsenbeek M, Kreuter M, Olson A, et al. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35(11):2015-24.
-
Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47.
-
Kolb M, Vašáková M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. 2019;20(1):57.
-
Nambiar AM, Walker CM, Sparks JA. Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians. Ther Adv Respir Dis. 2021;15:17534666211039771.
-
George PM, Spagnolo P, Kreuter M, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. 2020;8:925–54.
-
Kreuter M, Bendstrup E, Russell AM, et al. Palliative care in interstitial lung disease: living well. Lancet Respir Med. 2017;5(12):968-80.
-
Hagan TL, Xu J, Lopez RP, et al. Nursing’s role in leading palliative care: A call to action. Nurse Educ Today. 2018;61:216-9.
-
Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med. 2020;383(10):958-68.
-
National Institute for Health and Care Excellence. Idiopathic pulmonary fibrosis in adults: diagnosis and management (CG163). London: NICE, 2013.
-
Hoffmann-Vold A-M, Maher TM, Philpot EE, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. The Lancet Rheumatology. 2020;2(2):e71-e83.
-
Perelas A, Silver RM, Arrossi AV, et al. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. 2020;8(3):30420.
-
Wijsenbeek MS, Moor CC, Johannson KA, et al. Home monitoring in interstitial lung diseases [published online ahead of print, 2022 Oct 4]. Lancet Respir Med. 2022;S2213-2600(22)00228-4.
Related Items

Uppföljningsfrekvens vid ILD

